Full-sequence Computational Design and Solution Structure of a Thermostable Protein Variant.
Shah, P.S., Hom, G.K., Ross, S.A., Lassila, J.K., Crowhurst, K.A., Mayo, S.L.(2007) J Mol Biology 372: 1-6
- PubMed: 17628593
- DOI: https://doi.org/10.1016/j.jmb.2007.06.032
- Primary Citation of Related Structures:
2P6J - PubMed Abstract:
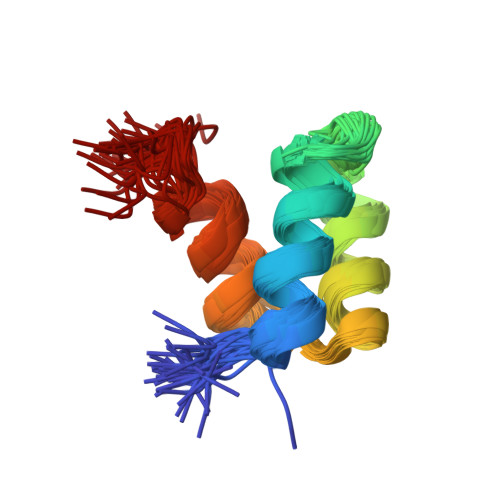
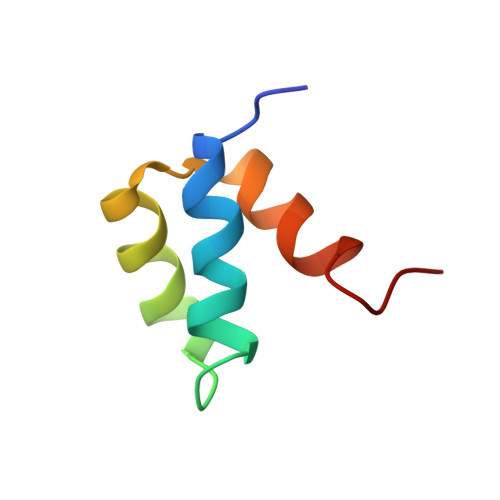
Computational protein design procedures were applied to the redesign of the entire sequence of a 51 amino acid residue protein, Drosophila melanogaster engrailed homeodomain. Various sequence optimization algorithms were compared and two resulting designed sequences were experimentally evaluated. The two sequences differ by 11 mutations and share 22% and 24% sequence identity with the wild-type protein. Both computationally designed proteins were considerably more stable than the naturally occurring protein, with midpoints of thermal denaturation greater than 99 degrees C. The solution structure was determined for one of the two sequences using multidimensional heteronuclear NMR spectroscopy, and the structure was found to closely match the original design template scaffold.
Organizational Affiliation:
Biochemistry and Molecular Biophysics Option, MC 114-96, California Institute of Technology, Pasadena, CA 91125, USA.